Navigating Assay Validation in Cell Therapy
Bringing cell therapies from the laboratory to patients isn’t just about innovation—it’s about trust. Every step of the journey, from early research to full-scale production, depends on the ability to measure, analyze, and understand these living therapies with confidence. At the core of this trust lies assay validation, the process that ensures each analytical method is precise, reliable, and capable of meeting the highest quality and regulatory standards.
At the beginning, analytical methods are exploratory, shaped by scientific discovery and initial feasibility studies. As development progresses, these methods are becoming more refined and more tightly controlled. By the time a therapy reaches GMP manufacturing, every assay must deliver consistent, high-quality data that ensures both regulatory approval and, most importantly, patient safety.
THE ANALYTICAL PROCESS JOURNEY
The analytical process unfolds in three key stages: Method development establishes a reliable foundation for the method, method qualification (or characterization) explores its performance and suitability, and method validation ensures it meets regulatory standards for its intended use.
It begins with R&D developing analytical methods tailored to the product’s characteristics and aligned with regulatory expectations. Quality Control (QC) is involved early on to assess feasibility and prepare for a smooth transfer. Once the method is sufficiently mature, it undergoes method qualification to verify key performance attributes such as accuracy and reproducibility under controlled conditions. At this stage, both Quality Control and Quality Assurance (QA) are involved. Subsequently, QC and QA oversee the method validation process, ensuring it meets regulatory standards.
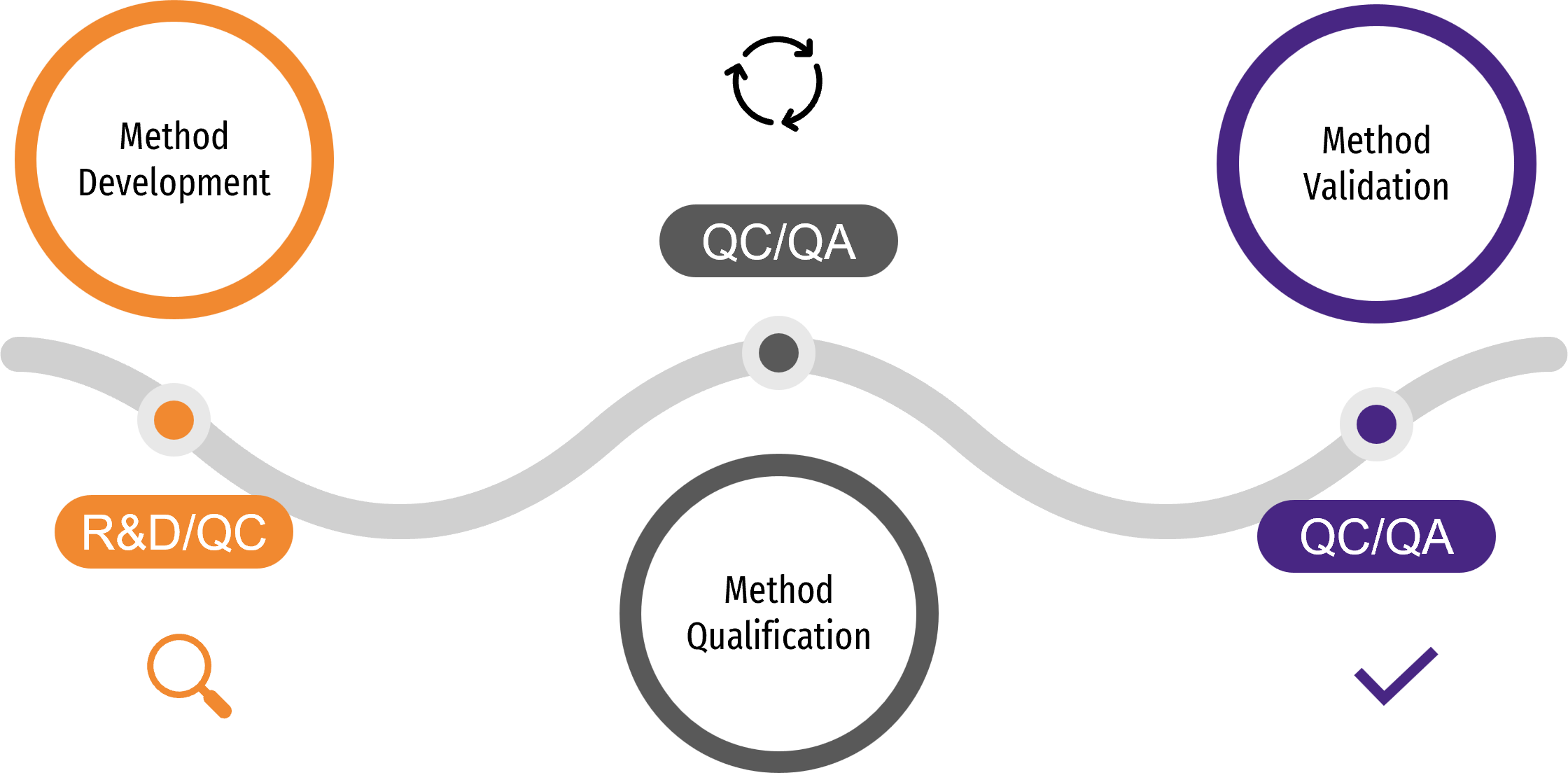
This analytical process follows the lifecycle of the product itself, evolving as the therapy progresses through its development stages. This alignment makes a phase-appropriate strategy essential, as it tailors the validation efforts to the specific needs and complexities of each stage. As development progresses, methods are refined, with extensive validation becoming mandatory for all of them to meet the demands of regulatory standards. Here are the key steps in the lifecycle of analytical methods:
1. Preclinical stage: Method Development
The development of analytical methods begins during the early preclinical research phases, where the methods used to assess cell therapies are often still in the experimental and exploratory stages. A key step in this process is defining an Analytical Target Profile (ATP), which forms the foundation for effective assay validation. An ATP is a predefined statement that outlines the purpose of an analytical method, its required performance characteristics, and performance criteria. It serves as a guiding framework to ensure that analytical methods are fit for purpose and capable of generating reliable and reproducible data. In the preclinical stage, the ATP is typically broad and flexible and evolving as development progresses. To effectively define an ATP, the process follows a structured approach:
Identify the Critical Quality Attributes (CQAs): The first step is to determine the key attributes that define the quality, safety, and efficacy of the therapy. These should be carefully selected to reflect their impact on product consistency and patient outcomes. These typically include lack of microbiological contamination, cell viability, identity, potency and purity. Early in development, some CQAs may not be fully defined, and their importance may evolve as more data is gathered.
Select appropriate analytical methods: The next step is to select analytical methods to measure CQAs accurately. Initially, it may not be possible to define all methods with precision, as they will evolve alongside the product. These will be adjusted as the product’s characteristics become clearer and more data is collected. The table below is a non-exhaustive list of key Critical Quality Attributes (CQAs) for cell therapies and the analytical methods used for their assessment:

Define performance characteristics: They include key aspects like precision, specificity, repeatability, sensitivity, and stability. For instance, a method to assess cell viability should have good precision (the ability to yield consistent results), specificity (the ability to measure only the intended parameter), and repeatability (the ability to reproduce results consistently across different trials). As the product progresses, these performance characteristics will become more refined, and the acceptance criteria for each will be adjusted accordingly.
2. Early Clinical Stage: Method Qualification
Upon submission of the Investigational Medicinal Product Dossier (IMPD), the primary focus shifts to evaluating the safety of your product. During this phase, your methods should undergo method qualification. However, methods related to safety, sterility, and microbial testing must already be validated following regulatory standards. Specific requirements from the EMA and FDA are detailed later in this article. At this stage, you begin quantitatively assessing the basic performance characteristics of your analytical methods.
For example, when measuring live cell concentration, it is common to establish an acceptance criterion for repeatability. At Cell-Easy, we apply a maximum variability of 10% at this phase. While the acceptance limits can remain relatively broad due to the exploratory nature of early clinical work, they will be progressively refined over time.
3. Late-Stage Development & Method Validation
As the product progresses into clinical trials, all methods should be validated following regulatory standards such as ICH Q2(R2). The validation process confirms that critical performance characteristics are well established and compliant with regulatory expectations. Full GMP-grade methods are mandatory during this phase.

4. Commercial Manufacturing
Once a cell therapy product receives marketing authorization, the final validated analytical methods are included in the Biological License Application (BLA) or Marketing Authorization Application (MAA). To ensure long-term assay reliability, continued performance monitoring is implemented throughout the product’s lifecycle.
REGULATORY LANDSCAPE – SOME GUIDELINES
Understanding regulatory guidelines is essential for ensuring compliance and streamlining cell therapy development. Regulatory agencies such as the FDA and EMA outline distinct yet complementary expectations that evolve throughout clinical development. Below are key regulatory expectations for method qualification and validation across different stages of development.
FDA Expectations
The FDA’s CMC guidance indicates that full analytical method validation is not required for initial IND submissions (Phase I) in cell therapy. However, test methods must be scientifically rigorous, well-controlled, and capable of ensuring specificity, sensitivity, and reproducibility. The FDA encourages the use of compendial methods where applicable and mandates qualification of safety-related assays before clinical trials. Additionally, assays detecting potential contaminants (e.g., residual cytokines, adventitious agents) should meet FDA sensitivity standards early in development.
EMA Expectations
The EMA aligns with the FDA on many principles but places greater emphasis on safety assay validation. According to EU GMP guidelines for ATMPs:
- Early-stage trials: Sterility and microbial testing must be validated, along with assays critical to patient safety (e.g., replication-competent virus detection for viral vector-based cell therapies).
- Throughout development: Full validation is not required, but methods must reliably assess critical quality attributes such as cell viability, impurity detection, and functional characterization. Potency assays should be validated before pivotal trials.
- Pivotal trials: Analytical methods for batch release and stability testing must be fully validated.
ASSAY VALIDATION: PARTNERING WITH CELL-EASY
Partnering with a CDMO for assay validation can be a strategic move to streamline your development process. By tapping into their expertise and infrastructure, you gain access to efficient methods and technologies that can help ensure your assays meet regulatory standards. This collaboration allows you to optimize your methods while managing risks and improving data integrity.
For any assistance or more information, feel free to contact us—we’re here to help guide you through every step of the process. Explore our services at www.cell-easy.com or contact us at info@cell-easy.com